1 Introduction
Table of contents
1.1 Challenges
In the 1980s, most non-protein-coding regions in the genome were thought to be ‘junk’ DNA with no functional purpose [1]. Nonetheless, during the last two decades, new classes of non-coding RNAs (ncRNAs) that have gene regulatory roles have been discovered within these ‘junk’ regions [2]. MicroRNAs (miRNAs) are one such new class of ncRNAs that have many important regulatory roles on a genome-wide scale [3]. Because of their importance, research on miRNAs has gained popularity in recent years (Fig. 1.1). Although many research efforts have revealed basic miRNA characteristic and regulation [3,4], there are still many challenges to identify the comprehensive characteristics and precise regulatory mechanism of miRNAs. This thesis covers three such challenges related to miRNA studies.
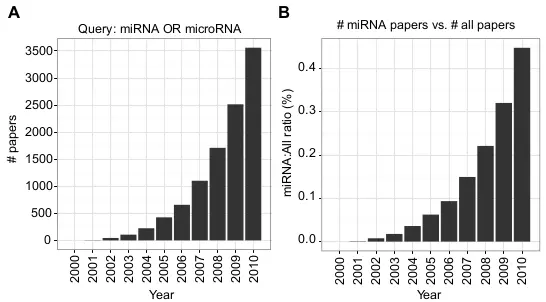
Figure 1.1. PubMed query.
Two figures show the trend of miRNA related papers as (A) the number of papers, and (B) the ratio to all papers in PubMed.
The first challenge is to identify accurate miRNA targets in animals. It is important to understand miRNA contributions to the genome-wide gene regulation, but there are several known obstacles related to indentifying miRNA targeRts in animals. Firstly, miRNAs are quite abundant [3], and one miRNA can potentially regulate many protein-coding genes [5]. In some cases, miRNAs bind their target mRNAs by base-pairing with only six nucleotides [5], which results in thousands of potential candidate genes influenced by one miRNA at a genome-wide level. Secondly, since miRNAs are expressed in a cell- or tissue-specific manner [6], one true positive miRNA target can be a false positive in a different cell or tissue type. Thirdly, the precise mechanism of miRNA binding process on its target mRNA is unknown [4]. Therefore, combinations of miRNA features are usually used to predict miRNA targets, but the combined effects of these features on miRNA targeting are unclear.
The second challenge is to interpret miRNA high-throughput data appropriately with high accuracy. Microarray, next generation sequencing, and quantitative proteomics are three major high-throughput technologies widely used for miRNA studies. Nonetheless, analyses of the data from these high-throughput technologies often give different interpretations regarding miRNA characteristics and regulation [7,8,9]. A major obstacle is that there are many factors involved in these analyses, but the main factors that cause these differences are unknown.
The third challenge is to indentify potential miRNA interactions with other ncRNAs. Although most miRNAs regulate genes at the post-transcriptional level, some miRNAs can also regulate transcription itself [10,11,12]. This transcriptional regulation seems to involve ncRNAs overlapping or interacting with the target gene promoters [13,14,15,16,17]. Many aspects of this miRNA regulation at the transcription level are poorly understood. Moreover, few experimental data are available for this miRNA regulation at the transcription level.
1.2 Goals
The main goal of this thesis is to reveal the characteristics and regulations of miRNAs by analyzing several different types of high-throughput data through bioinformatics approaches. To achieve this goal, I defined three sub-goals to solve the three challenges of miRNA studies described in the previous section.
The first sub-goal is to develop a miRNA target prediction algorithm with high accuracy. Most existing prediction algorithms focus on identifying individual target sites without considering multiple target sites. They do not include multiple target sites that possibly contribute to miRNA regulation. Moreover, most algorithms use strict filtering, such as filtering with evolutional conservation. Filtering can reduce false positive miRNA targets, but it potentially removes many true positive targets at the same time. Therefore, the aim of this sub-goal is to develop a model that can predict unbiased miRNA targets by considering multiple targets without filtering.
The second sub-goal is to analyze several different types of miRNA high-throughput technologies. The aim of this sub-goal is to reveal the characteristics of each technology and identify strong factors that cause inconsistent results between different types of experiments by statistical approaches.
The third sub-goal is to infer potential miRNA regulations outside of 3’ untranslated regions (UTRs) in general and interactions between miRNAs and ncRNAs in complex loci in particular. A complex locus is a region of DNA that contains multiple genes that have interactions between them or share common regulatory mechanisms [18]. Our hypothesis is that some miRNAs interact with ncRNA:mRNA pairs in complex loci. The aim of this sub-goal is to investigate this hypothesis of miRNA involvement in complex loci together with miRNA regulations outside of 3’ UTRs by computationally analyzing the data from high-throughput experiments.
In this thesis, these sub-goals are referred to in italic to clarify the relationship between parts of the text and their corresponding sub-goals if necessary.
-
First sub-goal: miRNA target prediction
-
Second sub-goal: miRNA high-throughput experiments
-
Third sub-goal: miRNA and other ncRNAs
1.3 Thesis structure
This thesis consists of eight chapters followed by five papers.
Chapter Two: Papers and their corresponding sub-goals. This chapter summarizes the five papers included in this thesis. It also relates them to each sub-goal.
Chapter Three: MicroRNAs and other non-coding RNAs. This chapter introduces the history, characteristics, and biological functions of miRNAs as well as some additional information about other ncRNAs.
Chapter Four: High-throughput biological experiments. This chapter focuses on three high-throughput technologies used in our research: microarray, next generation sequencing, and quantitative proteomics.
Chapter Five: Statistical tests and methods. This chapter starts with explaining basic statistical tests followed by applied statistical approaches used throughout in our research, such as non-parametric tests, resampling, and multiple comparison tests.
Chapter Six: Machine learning theory and Support vector machine. Support vector machine (SVM) is the main method used in the first sub-goal: miRNA target prediction. This chapter explains the theoretical background of SVM, data preparation and evaluation methods for SVM, as well as some other machine learning methods for comparison.
Chapter Seven: Computational implementation. Any state-of-the-art model or algorithm is ineffective without appropriate computational implementation. This chapter focuses on the computation implementations used in our research.
Chapter Eight: Future perspective. This chapter describes potential improvements of each sub-goal as future perspectives.
References
- Orgel LE, Crick FHC. Selfish DNA: the ultimate parasite. Nature 1980;284:604–7. https://doi.org/10.1038/284604a0.
- Wright MW, Bruford EA. Naming ’junk’: Human non-protein coding RNA (ncRNA) gene nomenclature. Human Genomics 2011;5:90. https://doi.org/10.1186/1479-7364-5-2-90.
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97. https://doi.org/10.1016/s0092-8674(04)00045-5.
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–33. https://doi.org/10.1016/j.cell.2009.01.002.
- Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research 2008;19:92–105. https://doi.org/10.1101/gr.082701.108.
- Wang L, Oberg AL, Asmann YW, Sicotte H, McDonnell SK, Riska SM, et al. Genome-wide transcriptional profiling reveals MicroRNA-correlated genes and biological processes in human lymphoblastoid cell lines. PLoS ONE 2009;4:e5878. https://doi.org/10.1371/journal.pone.0005878.
- Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature 2008;455:64–71. https://doi.org/10.1038/nature07242.
- Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008;455:58–63. https://doi.org/10.1038/nature07228.
- Wen J, Parker BJ, Jacobsen A, Krogh A. MicroRNA transfection and AGO-bound CLIP-seq data sets reveal distinct determinants of miRNA action. RNA 2011;17:820–34. https://doi.org/10.1261/rna.2387911.
- Place RF, Li L-C, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proceedings of the National Academy of Sciences 2008;105:1608–13. https://doi.org/10.1073/pnas.0707594105.
- Kim DH, Saetrom P, Snove O, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proceedings of the National Academy of Sciences 2008;105:16230–5. https://doi.org/10.1073/pnas.0808830105.
- Younger ST, Corey DR. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Research 2011;39:5682–91. https://doi.org/10.1093/nar/gkr155.
- Han J, Kim D, Morris KV. Promoter-associated RNA is required for RNA-directed transcriptional gene silencing in human cells. Proceedings of the National Academy of Sciences 2007;104:12422–7. https://doi.org/10.1073/pnas.0701635104.
- Morris KV. The emerging role of RNA in the regulation of gene transcription in human cells. Seminars in Cell & Developmental Biology 2011;22:351–8. https://doi.org/10.1016/j.semcdb.2011.02.017.
- Morris KV, Santoso S, Turner A-M, Pastori C, Hawkins PG. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genetics 2008;4:e1000258. https://doi.org/10.1371/journal.pgen.1000258.
- Schwartz JC, Younger ST, Nguyen N-B, Hardy DB, Monia BP, Corey DR, et al. Antisense transcripts are targets for activating small RNAs. Nature Structural & Molecular Biology 2008;15:842–8. https://doi.org/10.1038/nsmb.1444.
- Yue X, Schwartz JC, Chu Y, Younger ST, Gagnon KT, Elbashir S, et al. Transcriptional regulation by small RNAs at sequences downstream from 3′ gene termini. Nature Chemical Biology 2010;6:621–9. https://doi.org/10.1038/nchembio.400.
- Engström PG, Suzuki H, Ninomiya N, Akalin A, Sessa L, Lavorgna G, et al. Complex loci in human and mouse genomes. PLoS Genetics 2006;2:e47. https://doi.org/10.1371/journal.pgen.0020047.